سه شنبه, ۲۵ اردیبهشت, ۱۴۰۳ / 14 May, 2024
مجله ویستا
فناوری انفورماتیک و فناورینانو: قابلیتها، چالشها

● گرافیک کامپیوتری:
رشد گرافیک کامپیوتری محتملاً بزرگترین مولفه فناوریانفورماتیک در گستره وسیعی از شبیه سازیهای مولکولی است:
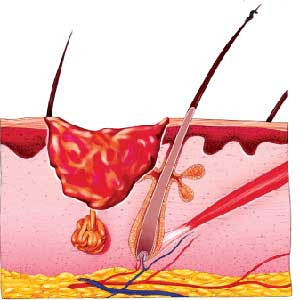
قابلیت تصویر سازی اهداف داروها( زمانی که ساختار آنها شناخته شده است) یا خانواده ترکیبی از آنالوگهای فعال و غیرفعال در حوزه مدلسازی، ضروری مینماید.(مثلاً در ساخت شیمیایی مواد) گرافیک کامپیوتری قدمتی از آغاز ترکیب صدا و رنگ به منظور شفاف سازی پیامها، تاکنون که با هدف توسعه دانش تصویرسازی کامپیوتری برای نمایش پروسههای شیمیایی و بیوفیزیکی، بکار گرفته می شود، دارد.
● «هندسه بعد»:
زمینه مهم دیگری که توسعه علمی آن اثری مهم،حداقل در شبیهسازی و مدلسازی در زیست – فیزیک و زیست – شیمی و صنعت داروسازی بر جای خواهد گذاشت، دانش«هندسه بعد» است .
انتقال این حوزه از ریاضیات محض بسوی علمشیمی و سیستمهای مولکولی بوسیله"Grippen" صورت گرفت." "Kuntz","Havel در اواخر دهه ۷۰ میلادی ( CrippenوHavel در ۱۹۸۸ میلادی) این تکنیک را در اندازهگیریهایNMR و QSAR بکار گرفتند. Crippen این پروسه را تا تحقق این علم برای مدلسازی بر هم کنش ماکرومولکولهای پیوندی، ادامه داد. (Crippen ۱۹۹۹)
● QSR/QSPR:
سومین زمینه مدلسازی شامل ارتباط میان دادههای آزمایشگاهی با همان خواص فیزیکی یا غیر آن، در قالب سیستم مدلسازی است. این روشها که موسوم به QSR/QSPR میباشند دارای تاریخچهای طولانی در مدلسازی سیستمهای بیولوژیک میباشند و اکنون نیز نقش مؤثری در تحلیل نتایج و استفاده از نتایج مدلسازی مولکولی در صنایع شیمی دارند.
یک مثال نمونه جهت استفاده از این تکنیکها این است که:
نمونهای را با یک سری از خواص فیزیکی در نظر بگیرید که میخواهید آن را بهینه کنید(سودمندی دارو، ارتباطات آنزیمی، مقاومت کششی یک پلیمر)
▪ چگونه آن را حل خواهید کرد:
در زمره تازهترین روشهای حل این مسأله که متعلق به "Hansch" می باشد (۱۹۷۱) مبتنی بر ارتباط فعالیتهای بیولوژیک مولکول در قالب پارامتر«آب گریزی» آن که به عنوان ثابت اکتاتل به آب، معرفی میشود.
این روش، در طول انرژی آزاد خطی در شیمی فیزیک زیستی قرار میگیرد. (یعنی: روابط Hammett(۱۹۳۵))، که انرژی آزاد(لگاریتم ثابت تعادل)، به نوع دیگری از انرژی یا خواص ماده که بر اساس انرژی آزاد سنجیده میشود، وابسته است.
همانظور که پیشنهاد شد، سادهترین چنین روشهائی به عنوان مدلهای خطی مطرحند و رگریسون خطی برای آن استفاده میشود. به همین ترتیب که مدل پیچیدهتر میشود، مدل به سمت رگریسون غیر خطی میل میکند(Kowalski ۱۹۸۴) و .(Andrea & Kalayeh۱۹۹۱)
به منظور توالی چنین مدلهائی، تا حصول روابطی میان خواص فیزیکی ماده ومیزان فعالیتهای آن می بایستی در مدلسازی به روش«شبکه عصبی» بکار گرفته شود.
علاوه بر «آب گریزی»، سایر پارامترهای مؤثر و مرتبط به عنوان متغیرهای مستقل در چنین مدلهائی، عبارتند از:
نسبت آرایش اتمها به نیروهای واندروالس و خواص الکترواستاتیک.
یک روش که در برگیرنده اطلاعات ۳ بعدی در قالب یک مدل باشد مانند «آنالیز میدانهای مولکولی تطبیقی که به اختصار COMFA خوانده میشود. COMFA: Comparative Molecular Field Analysis
که توسط" "Cramer،"Patterson" و" Bunce" در سال ۱۹۸۸ پایهگذاری شد این پارامترها و متغیرهای وابسته به آن عمومی می باشند و در بهینه سازی مدل مؤثرند. علاوه بر آن ارتباطات میان مولکولی را در جهت توجیه ساختار ماکرومولکول در ابعاد ۳ بعدی توجیه میکند. (به این معنی که نیروهای الکترواستاتیک و پتانسیلهای واندروالس محیط شده را لحاظ می کند.)
تکنیکهای فوق به نام QSAR خوانده می شود. QSAR: Quantitative Structure- Activity Relationship
واز مهمترین تکنیکهای فوق می توان به QSPR اشاره کرد.
QSPR: Quantitative Structure – Properties Relationship
که این تکنیک زمانی که بهینهسازی تعدادی از خواص مکانیکی بر اساس فعالیتشان مطلوب باشد، مورد توجه قرار میگیرد("Katrizky", "Lobanov"&" Karelson" ۱۹۹۵) .
یکبار دیگر، میتوان این عمل را در زمینه مدلهای خطی و غیرخطی انجام داد و میتوان خواص متعددی از مجموعهای از مولکولها را در چنین مدلی لحاظ کرد. بنابراین برای نتیجهگیری خواص الکترواستاتیک یا خواص الکترونی ماده، به منظور آزمایش اینکه آیا آنها واقعاً دارای چنین روابطی هستند، مکانیک کوانتوم محاسباتی استفاده میشود. از آنجائیکه هدف نهائی دستیابی به ساختار دقیق الکترونی ماده نمیباشد و دقت کمتر و کارآئی بیشتر مطلوب است، (روشهای شبه تجربی) روشهای مکانیک کوانتوم برای چنین اهدافی مطلوب است.
● استفاده از«شبیه سازی» در تعیین ساختار:
استفاده از «دینامیک مولکولی» در شناسائی ساختار به کمک X-RAY و تعیین ساختار NMR یکی از مهمترین و موفق ترین هدفها در مدلسازی مولکولی است. این روش بصورت کاملاً قاطعانهای در تعیین ساختار کریستالی پروتئین و ساختار زئولیتهای جدید مؤثر است. از آنجائیکه دادههایX-RAY در کریستالوگرافی ماکرومولکولی جهت تعیین ساختار اتمی با دقت بالا کافی نمیباشد، در مقابله با چنین روشی در ساختار کریستالی "کوچک – مولکول"، استفاده از ساخت مولکول و تکنیکهای شبیه سازی آن برای پر کردن این خلاء الزامی است.
برنامه XPLOR که توسط" Kuriyan"," Karplus", "Brunger" که در سال ۱۹۸۷، نوشته شد، در جهت توسعه و ترکیب دانش دینامیک مولکولی و تکنیکهای X-Ray گام بر میداشت و یک نمونه مناسب در راستای تشریح روش ترکیب این دو شاخه بود. در این متدولوژی، ساختار اولیه با استفاده از یک تابع که نتیجه گرفته شده از تفاوتهای میان محاسبات و بازخوردهای آزمایشگاهی برداشت شده، و تابع انرژی مولکولی مکانیکی است، تعیین میشود. با استفاده از دینامیک مولکولی و دمای بالا، و جابجائی ساختاری در طی یک کمینه موضعی با تغییر در وزن انرژی مولکولی مکانیکی و سوار کردن بازخوردهای آزمایشگاهی، میتوان سیستم را بسوی سازهای با استریوشیمی مطلوب و حداکثر دقت که مبتنی بر نتایج آزمایشگاهی است رهنمون شد.
(بر اساس مشاهدات ما، یک گروه مدل ساز مولکولی، بصورت غیر همزمان، و یک گروه کریستالوگراف شناسی تجربی، اعلام کردند که مدلسازی مولکولی در طراحی دارو، به اندازه کریستالوگرافی به کمک دینامیک مولکولی مؤثر نمیباشد.)
اهمیت مدلسازی به کمک دینامیک مولکولی در تعیین ساختار سیستمهای ماکرومولکول به کمک NMR، نمود بیشتری پیدا میکند، به ویژه آنجائیکه، تعداد دادههای هندسی آزمایشگاهی مشخصاً کمتر از تعداد دادههای کریستالوگرافی است("Case","Wiley" & " Chechester" ۱۹۹۸) بنابراین استفاده از فناوریهای متنوع در جهت ساخت مدل ۳ بعدی ماده، نظیر تکنولوژیهای « هندسه بعد »یا« دینامیک مولکولی»، نقش مهمی در NMR سازه در مقیاس ماکرو مولکولی بازی میکند.
● تکیه گاهی به درون اهداف بیولوژیک:
اگر ساختار ماکرومولکولی هدف معلوم باشد، ولی ارتباطات و پیچیدگیهای آن معلوم نباشد، لازم است تا روشهای محاسباتی که با دقت بالائی ساختار و پیچیدگیهای پروتئین را پیش بینی کنند، مد نظر قرار گیرد نه فقط برای یک پیوند بلکه برای تمام مجموعه پیوندها.
برنامه DOCK ("Zou", "Sun", "Kuntz" ۱۹۹۹) و برنامهFLEXX ("Kramer" et al. ۱۹۹۹) ، از مجموعه برنامه هایی است که توانایی لحاظ کردن مجموعه ای در حدود ۰۰۰/۱۰۰ پیوند به منظور شبیه سازی کامل یک ماکرومولکول هدف، را دارد.
در سوی دیگر این طیف، برنامه هایی نظیر برنامه("Rao" & "Olsen" ۱۹۹۹) ، موسوم به AUTODOCK میباشند که گستره وسیعی از پیوندهای بین مولکولی را در یاخته ها با استفاده از روش مونت کارلو شبیه سازی میکنند.نیزروشهائی جهت لحاظ کردن تمام ترکیبات و حالات حاکم بر پیوندها، توسعه یافته است(Kicketal.۱۹۹۷) که این روشها تأثیر خاصی در شبیه سازی ساختارها دارند.
● زیست انفوماتیک و شیمی انفورماتیک:
توانائی در جهت آنالیز، ساخت و جستجو در تمام رشته های ژنتیکی ارگانیزمهای متعدد، هدف عالی زیستانفورماتیک است. تعمیم همه انواع آنالیزها در قالب بانک اطلاعاتی های بزرگ گردآوری شده از مولکولهای زیستی، هدف عالی شیمی انفوماتیک است.
هدف زیست انفورماتیک این است که کشف ژن، تابع ژن، طراحی دارو، توسعه دارو، را در قالب الگویی مناسب ترکیب کند.
زیست انفورماتیک و شیمی انفورماتیک را باید بخشی از پروسه کلانتری در نظر گرفت که مبتنی بر فهرست گیری، ذخیره سازی، دستکاری و آنالیز گستره وسیعی از اطلاعات ژنها، توصیف ژنها و پروتئینهای سازنده است و اثری که هر یک از ریز مولکولها ممکن است بر DNA یا RNA داشته باشد را اندازه گیری می کند.
زیست انفورماتیک و شیمیانفورماتیک دارای نظمی کلان ومهم میباشند. شیمی محاسباتی و مدلسازی بخشی از حوزه دانش انفورماتیک هستند.
● علوم کامپیوتر:
بسیاری از محاسبات در حوزه مولکولی نیازمند منابع کامپیوتری قدرتمند است و عملاٌ توسعه این محاسبات نیازمند توسعه دانش کامپیوتر است به همین ترتیب حوزه آنالیز اطلاعات نیز ریشه در این علوم دارد.
نرم افزارهای مبتنی بر محاسبات برداری و محاسبات موازی در ۱۵ سال آینده اثری مهم در حوزه فناوری بر جا خواهند گذاشت. خوشههای کامپیوتری درجهت توسعه محاسبات موازی روبه توسعه خواهند گذاشت و حوزه صنعت و تحقیقات را متحول خواهد کرد.
http://www.technologyworld.blogsky.com
منابع :
۱) Andrea, T., and H. Kalayeh. ۱۹۹۱. J. Med. Chem. v. ۳۴. p. ۲۸۲۴.
۲) Brunger, A.T., J. Kuriyan, and M. Karplus. ۱۹۸۷. Science. ۲۳۵, ۴۵۸.
۳) Case, D., J. Wiley, and Chichester. ۱۹۹۸. NMR refinement. Encyclopedia of Computational Chemistry. P.v.R. Schleyer(ed.). p. ۱۸۶۶.
۴) Cramer, R.D., D.E. Patterson, and J.D. Bunce. ۱۹۸۸. J. Amer. Chem. Soc. v. ۱۱۰. p. ۵۹۵۹.
۵) Crippen, G.M., and T.F. Havel. ۱۹۸۸. Distance Geometry and Molecular Conformations. Research Studies Press. Wiley.New York.
۶) Ferrin, T.E., and T.E. Klein. ۱۹۹۸. Computer graphics and molecular modeling. Encyclopedia of Computational
۷) Hammett, L.P. ۱۹۳۵. Chem. Rev. v. ۱۷. ۱۲۵.
۸) Hansch, C. ۱۹۷۱. Drug Design, E.J. Ariens (ed.). Academic Press. New York. vol. ۱, chapter ۲.
۹) Katrizky, A., V. Lobanov, and M. Karelson. ۱۹۹۵. Chem. Soc. Rev. v. ۲۴. ۲۷۹.
۱۰) Kowalski, B. (ed.). ۱۹۸۴. Chemometrics.Mathematics and Statistics in Chemistry. D. Reidel Publishing Co. Dordecht.
۱۱) Joliffe, I.T. ۱۹۸۶. Principal Component Analysis. Springer-Verlag. New York.
۱۲) Kramer, B., G. Metz, M. Rarey, and T. Lengauer. ۱۹۹۹. Med. Chem. Res. ۹. ۴۶۳.
۱۳) Kick, E.M., E.M.D.C. Roe, A.G. Skillman, G.C. Liu, T.J.A. Ewing, Y.X. Sun, I.D. Kuntz, and J.A. Ellman. ۱۹۹۷.
۱۴) Langridge, R., T.E. Ferrin, I.D. Kuntz, and M.L. Connolly. ۱۹۸۱. Science. ۲۱۱. p. ۶۱۱.
۱۵) Rao, M.S., and A.J. Olson. ۱۹۹۹. Proteins.Structure, Function and Genetics .۳۴. ۱۷۳.
۱۶) Zou, X.Q., Y.X. Sun, and I.D. Kuntz. ۱۹۹۹. J. Amer. Chem. Soc. ۱۲۱. ۸۰۳۳.
۱۷) Thayer, A.M. ۲۰۰۰. Bioinformatics for the masses. Chem. Eng. News. ۱۹.
منابع :
۱) Andrea, T., and H. Kalayeh. ۱۹۹۱. J. Med. Chem. v. ۳۴. p. ۲۸۲۴.
۲) Brunger, A.T., J. Kuriyan, and M. Karplus. ۱۹۸۷. Science. ۲۳۵, ۴۵۸.
۳) Case, D., J. Wiley, and Chichester. ۱۹۹۸. NMR refinement. Encyclopedia of Computational Chemistry. P.v.R. Schleyer(ed.). p. ۱۸۶۶.
۴) Cramer, R.D., D.E. Patterson, and J.D. Bunce. ۱۹۸۸. J. Amer. Chem. Soc. v. ۱۱۰. p. ۵۹۵۹.
۵) Crippen, G.M., and T.F. Havel. ۱۹۸۸. Distance Geometry and Molecular Conformations. Research Studies Press. Wiley.New York.
۶) Ferrin, T.E., and T.E. Klein. ۱۹۹۸. Computer graphics and molecular modeling. Encyclopedia of Computational
۷) Hammett, L.P. ۱۹۳۵. Chem. Rev. v. ۱۷. ۱۲۵.
۸) Hansch, C. ۱۹۷۱. Drug Design, E.J. Ariens (ed.). Academic Press. New York. vol. ۱, chapter ۲.
۹) Katrizky, A., V. Lobanov, and M. Karelson. ۱۹۹۵. Chem. Soc. Rev. v. ۲۴. ۲۷۹.
۱۰) Kowalski, B. (ed.). ۱۹۸۴. Chemometrics.Mathematics and Statistics in Chemistry. D. Reidel Publishing Co. Dordecht.
۱۱) Joliffe, I.T. ۱۹۸۶. Principal Component Analysis. Springer-Verlag. New York.
۱۲) Kramer, B., G. Metz, M. Rarey, and T. Lengauer. ۱۹۹۹. Med. Chem. Res. ۹. ۴۶۳.
۱۳) Kick, E.M., E.M.D.C. Roe, A.G. Skillman, G.C. Liu, T.J.A. Ewing, Y.X. Sun, I.D. Kuntz, and J.A. Ellman. ۱۹۹۷.
۱۴) Langridge, R., T.E. Ferrin, I.D. Kuntz, and M.L. Connolly. ۱۹۸۱. Science. ۲۱۱. p. ۶۱۱.
۱۵) Rao, M.S., and A.J. Olson. ۱۹۹۹. Proteins.Structure, Function and Genetics .۳۴. ۱۷۳.
۱۶) Zou, X.Q., Y.X. Sun, and I.D. Kuntz. ۱۹۹۹. J. Amer. Chem. Soc. ۱۲۱. ۸۰۳۳.
۱۷) Thayer, A.M. ۲۰۰۰. Bioinformatics for the masses. Chem. Eng. News. ۱۹.












































































































































































نمایندگی زیمنس ایران فروش PLC S71200/300/400/1500 | درایو …
دریافت خدمات پرستاری در منزل
پیچ و مهره پارس سهند
تعمیر جک پارکینگ
خرید بلیط هواپیما
ایران آمریکا شورای نگهبان رهبر انقلاب مجلس مجلس شورای اسلامی صادق زیباکلام حجاب دولت سیزدهم انتخابات مجلس انتخابات مجلس دوازدهم
هواشناسی پلیس تهران بارش باران قوه قضاییه شهرداری تهران قتل سیل سلامت سازمان هواشناسی زلزله وزارت بهداشت
مسکن خودرو بازار خودرو قیمت دلار قیمت خودرو سایپا قیمت طلا بورس بانک مرکزی حقوق بازنشستگان گاز دلار
همایون شجریان محمد رسول اف نمایشگاه کتاب تهران کتاب نمایشگاه بینالمللی کتاب تهران سینمای ایران دفاع مقدس تئاتر تلویزیون رضا عطاران سریال
وزارت علوم دانشگاه تهران
اسرائیل جنگ غزه رژیم صهیونیستی غزه فلسطین حماس روسیه افغانستان طوفان الاقصی نوار غزه اوکراین ترکیه
استقلال فوتبال فولاد خوزستان پرسپولیس لیگ برتر فولاد مهدی طارمی رئال مادرید باشگاه استقلال لیگ برتر ایران لیگ برتر فوتبال ایران بازی
هوش مصنوعی فناوری گوگل تبلیغات شفق قطبی نوآوری ایلان ماسک ناسا اپل
زیبایی رژیم غذایی شیر فشار خون درمان ناباروری واکسن