شنبه, ۱ اردیبهشت, ۱۴۰۳ / 20 April, 2024
مجله ویستا
سندرم بروگادا
۱) بلوک شاخه راست
۲) ST- در V۱-V۳
۳) مرگ ناگهانی به صورت سنکوپ یا Sudden death در بیمارانی که قلب طبیعی دارند.
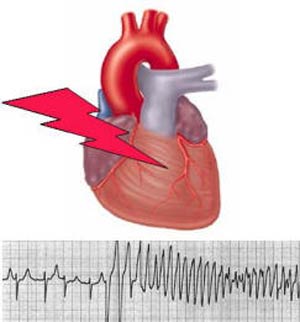
حوادث سنکوپ و مرگ ناگهانی به وسیلهی vt یا vf ایجاد میشوند. این آریتمیها بدون هشدار و به صورت ناگهانی ایجاد میشوند در حالی که ریتم بیمار سینوسی است و فاصلهی QT طولانی نمیشود.
فقط در موارد بسیار اندک، قبل از VT، نوسانات کوتاه و بلند ایجاد میشود، یافتهیی که در آریتمیهای دیگر نظیر Torsade de points در سندرم QT طولانی بسیار شایع است. قبل از آن، افزایش سرعت ضربان قلب، همانند تاکیکاردی بطنی پلی مرفیک وابسته به کاتکول آمین، وجود ندارد.
● تاریخچه:
اولین بار این سندرم در سال ۱۹۸۶ مشاهده شد. بیمار یک پسر سه ساله از لهستان بود. او به دفعات دچار این حمله شده و به وسیلهی پدرش احیاء شده بود. خواهر کوچکترش در سن دو سالگی پس از حملات متعدد به طور ناگهانی فوت شده بود.
۸ مورد دیگر از این سندرم درسال ۱۹۹۲ نیز گزارش شد. سپس در سراسر جهان نمونههایی مانند این بیماران گزارش گردید. کشف اخیر غیر طبیعی بودن ژنتیک این سندرم را مشخص کرد. در مرحلهی ابتدایی اختلالات الکتریکی این بیماری یک مرحلهی مهم در پیشگیری و درمان مؤثر برای جلوگیری از مرگ ناگهانی بیماران با ساختار قلب طبیعی میباشد.
در سال ۱۹۸۰، CDC (مرکز کنترل بیماریها) در آتلانتا، یک عارضهی بالای غیرطبیعی از مرگ ناگهانی را در مردان جوان از جنوب شرق آسیا گزارش کرد.
● شیوع بیماری:
این سندرم، شایعترین علت مرگهای ناگهانی در افراد جوان کمتر از پنجاه سال در جنوب آسیا است که سابقهی ناراحتی قلبی ندارند. در بعضی افراد نوار قلبی ممکن است به طور تیپیک (RBBB؛ تغییرات ST در V۱-V۳) بروز کند. اما نوار سایر افراد ممکن است طبیعی باشد و یا گاهی به طور متناوب غیر عادی شود.
الکتروکاردیوگرام غیر عادی در این سندرم ممکن است به دنبال تجویز «فلسیانید» یا «پروکایین آمید» « آجمالین» آشکار شود. بنابراین تست داروشناسی اختصاصی است و برای بیمارانی که دارای تاریخچهیی از سنکوپ با منشاء ناشناخته یا vf نامعلوم میباشند، توصیه میشود.
متأسفانه این آزمایشها به صورت روتین انجام نمی شوند و بدون تردید این یکی از دلایلی است که سبب عدم تشخیص به موقع این بیماری میشود. در فرانسه و ژاپن یک مورد از هزار الکتروکاردیوگرام در افراد بزرگسال ظاهراً سالم دارای این سندرم میباشند. این سندرم به طور متوسط، سبب مرگ ناگهانی در سن ۳۵ تا ۴۵ سالگی میشود. همچنین در سن ۶ ماهگی و ۷۴ سالگی نیز این سندرم مشاهده شده است.
● تظاهرات بالینی:
این سندرم با حملات vt پلی مرفیک یا vf مشخص میشود، وقتی حملهی تمام میشود، بیمار دچار حمله سنکوپ میشود. وقتی حمله ادامه پیدامیکند خود به خود قلب ضربه میبیند و دچار وقفه و در نتیجه مرگ ناگهانی میشود، بنابراین طیف بروز علائم گسترده است.
در یک سر آن اشخاصی هستند که بدون علامتاند و در سر دیگر آن افرادی که دچار مرگ ناگهانی میشوند. اشخاصی که الکتروکاردیوگرام آنها طبیعی است در ضمن آزمایشهای روتین شناسایی میشوند. در سایر بیماران، در ضمن غربالگری از طریق الکتروکاردیوگرام، بعد از مرگ ناگهانی در یکی از اعضای خانواده شناسایی میشوند.
● تشخیص:
تشخیص سندرم، آسان است، از طریق الکتروکاردیوگرافی دراز مدت در بیمارانی که شکل تیپیک این الکترو را دارند ( ST-در V۱-V۳ و RBBB) و همچنین تاریخچهیی از مرگ ناگهانی یا سنکوپ به دنبال vt پلی مرفیک در اعضای خانواده میباشد. تغییرات نوار در این سندرم وابسته به تداخل سیستم اتونوم و تجویز داروهای آنتیآریتمی میباشد:
داروهای تحریک کنندهی آدرنرژیک سبب کاهش صعود ST میشوند، در حالی که داروهای تحریک کنندهی واگ باعث بدتر شدن آن میشوند. داروهای کلاس ۳ و Ic و Ia و تب باعث افزایش صعود قطعه ST میشوند. ورزش در برخی بیماران، سبب کاهش صعود قطعهی ST میشود، اما در دیگران سبب افزایش صعود قطعهی ST میشود (بعد از ورزش، وقتی که درجه حرارت بدن افزایش مییابد).
وقتی ضربان قلب کاهش مییابد، صعود قطعه ST افزایش مییابد. وقتی که ضربان قلب تند میشود صعود قطعه ST کاهش مییابد، هر چند که خلاف این هم ممکن است مشاهده شود. بیماران دچارسنکوپ ناشناخته باید با داروهای آنتیآریتمی تحت بررسی قرارگیرند تا احتمال این سندرم به عنوان یکی از علل آریتمی بطنی و سنکوپ رد شود.
● پیش آگهی و درمان:
پیش آگهی این سندرم، وقتی بدون درمان گذاشته شود، خیلی وخیم است. یک سوم بیماران دچار سنکوپ یا تحت احیای قلبی ریوی، حملات vt پلی مرفیک جدید در طی ۲ سال بعد را داشتهاند.
متأسفانه پیش آگهی افراد بدون علامت همراه با الکتروکاردیوگرام تیپیک نیز خیلی بد است. یک سوم افرادی که هیچ سابقهی بیماری قلبی ندارند دچار اولین vt پلی مرفیک یا vf در طی ۲ سال بعد شدهاند. این علائم آخر (vt-vf) برای طرح درمان این بیماران خیلی مهم است: برای اینکه داروهای آنتیآریتمی (آمیودارون یا بتابلوکرها) از مرگ ناگهانی قلبی جلوگیری نمیکند، فقط کاشتن دفیبریلاتور بطنی (ICD) مؤثر و لازم میباشد و آریتمیهای بطنی را درمان میکند، وقتیکه دفیبریلاتور بطنی در قلب کاشته میشود، میزان مورتالیته در بیماران با سندرم بروگادا به ۰% با پیگیری ۱۰ ساله این بیماران میرسد. این جای تعجب نیست، چون این بیماران جوان هستند و معمولاً بیماریهای دیگری ندارند و ساختار قلب آنها نرمال میباشد و بیماری کرونر قلب ندارند، این بیماران نارسایی قلبی و اشکالات ایسکمی قلبی نیز ندارند، بنابراین کاندید ایدهآلی برای درمان با ICD میباشند.
بررسی معاینات بالینی و الکتروکاردیوگرافی از ۲۰ بیمار ایرانی (۱۵ مرد با میانگین سن ۹±۴۲ که سندرم بروگادا داشتند، انجام شد. الکتروکاردیوگرام غیر طبیعی (به صورت خود به خودی در۶ مورد و به دنبال تحریک در ۱۴ مورد) در ۵ بیمار علامتدار و ۱۵ مورد بیماران بدون علامت شناسایی شد. آریتمی بطنی در ۴۰% از بیماران بدون علامت ایجاد شد.
اختلاف مهمی در سن، جنس و PR اینتروال ST- ، یا اینتروالHV بین بیماران وجود نداشت. در ۸ بیمار دفیبریلاتور بطنی کاشته شد. در طول ۲±۱۶ ماه پیگیری، یک بیمار علامتدار، به طور مناسب تحت درمان با دستگاه شوک کاشتنی قرارگرفت. هیچیک از بیماران بدون علامت و بیماران دارای نوار غیرطبیعی خود به خودی دچار حملهی قلبی نشدند. اطلاعات الکتروفیزیولوژیک نقشی در پیشبینی تحریک برنامهریزی شده، ندارند.
● منشای الکتروکاردیوگرام و آریتمی بطنی:
روشن نیست در این سندرم RBBB، واقعی است و یا به علت دپلاریزاسیون زودرس اپیکاردی بطنی راست میباشد. علائم بالینی و الکتروفیزیولوژی نشان میدهد هر دو احتمال ممکن است وجود داشته باشد، بعضی از الکتروکاردیوگرامها به طور واضح یک RBBB را بعد از طبیعی شدن قطعهی ST نشان میدهد. تقریباً ۱۰% بیماران با این سندرم، با AF ناگهانی مراجعه میکنند، امکان دارد نقص ژنتیکی در بعضی از بیماران، منجر به تاکیکاردی بطنی و VF شود.
● ارتباط با سایر سندرمها:
امکان دارد بین این سندرم وسایر سندرمها، مخصوصاً دیس پلازی بطن راست ارتباطی وجود داشته باشد.اخیراً، غیر طبیعی بودن ژنتیکی سندرم بروگادا را کشف کردهاند، به این ترتیب که ایرادی در ژن SCNSA واقع روی کروموزوم ۳ وجود دارد . در حالی که در دیسپلازی بطن راست، ژن معیوب روی سایر کروموزومها قرارمیگیرد.
به رغم این بررسیها، ممکن است در بعضی از بیماران، دو شکل متفاوت بیماری ظاهر شود. در برخی مواقع الکتروکاردیوگرام مشابه سندرم بروگادا وجود دارد، اما به وسیلهی این سندرم ایجاد نمیشود (سندرم بروگادای کاذب).
«ایکوگوش» و همکارانش بررسی خوبی را روی یک بیماردچار سندرم «ولف پارکینسون وایت» انجام دادند که به وسیلهی کاتتری که دارای فرکانس رادیویی بود، تحت درمان قرار گرفته بود، بیمار بعد از دو سال به طور ناگهانی فوت کرد. آنالیز الکتروکاردیوگرام این مورد نشان میداد که بیمار علاوه بر«ولف پارکینسون» مبتلا به سندرم بروگادا نیز بود.
بنابراین اوهمزمان مبتلا به دوسندرم بود. پس این احتمال وجود دارد که بعضی از بیماران همزمان دچار دیسپلازی بطن راست و سندرم بروگادا باشند. کشف اخیر نشان میدهد که نقص ژنتیکی، عامل ایجاد این سندرم است.
این سوال مطرح شد که امکان دارد بیماری «شاگاس» سبب ایجاد صدمه روی کانال سدیم شود.گرچه تفاوتهای اساسی بین بیماری«شاگاس» وسندرم «بروگادا» وجود دارد، ولی در نهایت هر دو به مرگ ناگهانی در فرد منجر میشود. آسیب کانال سدیم در اثر عفونت و ایمونولوژیک دربیماری «شاگاس»ایجاد می شود، ولی در سندرم بروگادا جنبهی ژنتیک دارد واختلال در هدایت و رپولاریزاسیون، هدایت الکتریکی به هم میخورد و باعث ایجاد VF میشود.
سهیلا امیرسلیمانی
کارشناس پرستاری
کارشناس پرستاری
منبع : هفته نامه پزشکی امروز




همچنین مشاهده کنید

































































































نمایندگی زیمنس ایران فروش PLC S71200/300/400/1500 | درایو …
دریافت خدمات پرستاری در منزل
pameranian.com
پیچ و مهره پارس سهند
خرید میز و صندلی اداری
خرید بلیط هواپیما
گیت کنترل تردد
ایران اسرائیل حجاب اصفهان ایران و اسرائیل گشت ارشاد حمله ایران به اسرائیل حسین امیرعبداللهیان استان اصفهان دولت وعده صادق جنگ ایران و اسرائیل
تهران سیل وزارت بهداشت هواشناسی شهرداری تهران قتل فضای مجازی زلزله سازمان هواشناسی سیلاب قوه قضاییه آموزش و پرورش
قیمت دلار قیمت طلا خودرو قیمت خودرو دلار بازار خودرو بانک مرکزی چین ایران خودرو حقوق بازنشستگان تورم قیمت سکه
سعدی سینمای ایران تئاتر تلویزیون فیلم سینمایی روز سعدی سینما موسیقی سریال کتاب دفاع مقدس بازیگر
سازمان سنجش مغز
رژیم صهیونیستی غزه عراق فلسطین جنگ غزه حماس روسیه سازمان ملل امیرعبداللهیان ترکیه اسراییل شورای امنیت
فوتبال استقلال پرسپولیس شمس آذر قزوین تراکتور باشگاه استقلال صنعت نفت آبادان لیگ برتر لیگ برتر فوتبال ایران لیگ برتر ایران لیگ قهرمانان اروپا رئال مادرید
سرطان فناوری اینترنت ناسا هوش مصنوعی گوگل سامسونگ تبلیغات تلگرام اپل
حمله قلبی کلسترول فشار خون گیاهان دارویی پیاده روی